Julien Allali
MiGaL Tutorial
MiGaL Tutorial: Example, comparison of two 16S
compute scores:
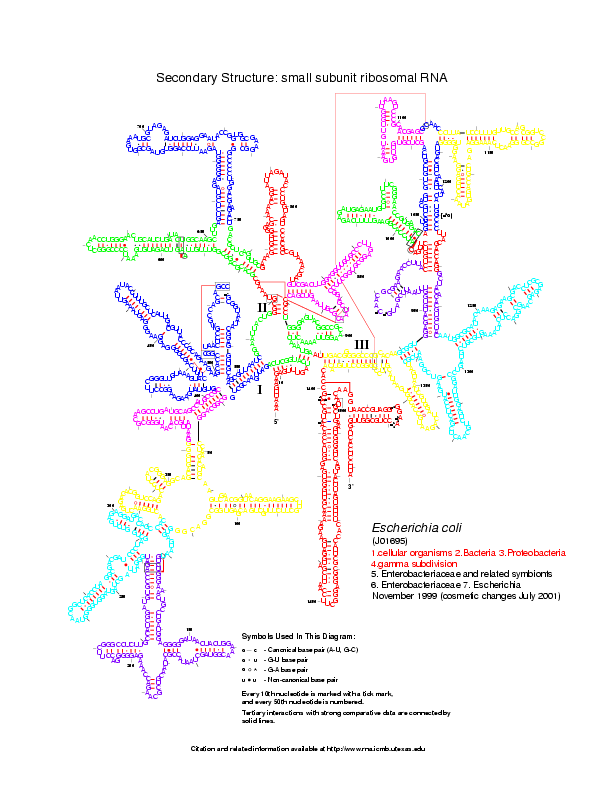
For this example, we use two RNA secondary structures coming from the Gutell Lab Comparative RNA web site: Bacillus subtilis 16S and Escherichia coli 16S.
To compare these structures, we used ''migal'':
migal -M d.16.b.B.subtilis.bpseq d.16.b.E.coli.bpseq
The program asks you Is helix number 0 a pseudo-k?. The
secondary structure in file ''d.16.b.B.subtilis.bpseq'' contains 106
helices listed
by the program. Some of these helices contain pseudo-knots which are
not supported by
migal. One therefore has to tell which helices must be broken so that
there are no
pseudo-knots. Here we say 'y' for helices 0, 38, 42 of the first
structure and for helices
0, 36, 42 and 46 of the second structure.
Then, the program computes eight values, two values for each of the four levels. As described in the paper presented at SPIRE'05 (see research section), MiGaL works with a four levels representation of the secondary structure. Each level is a tree:
- Level 0: multiloop network
- Level 1: stems network (=level 0+hairpin loops)
- Level 2: helices network (=level 1+internal loops and buldges)
- Level 3: base pairs and unpaired bases.
For each of these four layers, we have two values. The first is the constrained distance value (which is not really a distance because of the constraints). This value is the value of the edit distance with fusions with the contraints imposed by previous levels. This value represents the number of mismatches, insertions and deletions made during the computation. The second value is equal to the first value divided by the cost of the deletion of the first tree plus the cost of insertion of the second tree. Thus, the second value is between 0 and 1 and represents something like the percentage of mismatches/indels made during the edition.
ouput the alignment:
To obtain the alignment of the sequence resulting from the comparison, use the '-a' option:
migal -a alignment -M d.16.b.B.subtilis.bpseq d.16.b.E.coli.bpseqThe file alignment contains the alignment of the two sequences.
view the result:
MiGaL knows some Postscript formats. Thus, you can specify the postscript files of the first structure and of the second one (or of just one of them) to obtain a colored version of these postscripts:
migal --ps1 d.16.b.B.subtilis.ps --ps2 d.16.b.E.coli.ps -M
d.16.b.B.subtilis.bpseq d.16.b.E.coli.bpseqTwo new files are created: d.16.b.B.subtilis.ps_MiGaL.ps and d.16.b.E.coli.ps_MiGaL.ps which are coloured from the computation:
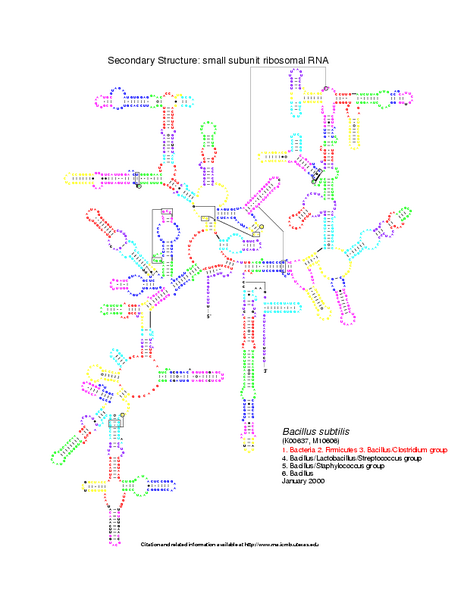

Note that MiGaL knows the Postscript created by RNAplot
(Vienna package)
with the --post "" option.
If you want to see to result of the comparison at each level you can use
the option --all-ps:
migal --ps1 d.16.b.B.subtilis.ps --ps2 d.16.b.E.coli.ps -M
d.16.b.B.subtilis.bpseq d.16.b.E.coli.bpseq --all-ps
This create 6 more files: d.16.b.B.subtilis.ps_MiGaL_0.ps, d.16.b.B.subtilis.ps_MiGaL_1.ps, d.16.b.B.subtilis.ps_MiGaL_2.ps and d.16.b.E.coli.ps_MiGaL_0.ps, d.16.b.E.coli.ps_MiGaL_1.ps, d.16.b.E.coli.ps_MiGaL_2.ps:
Level 0: the multiloop network comparison:
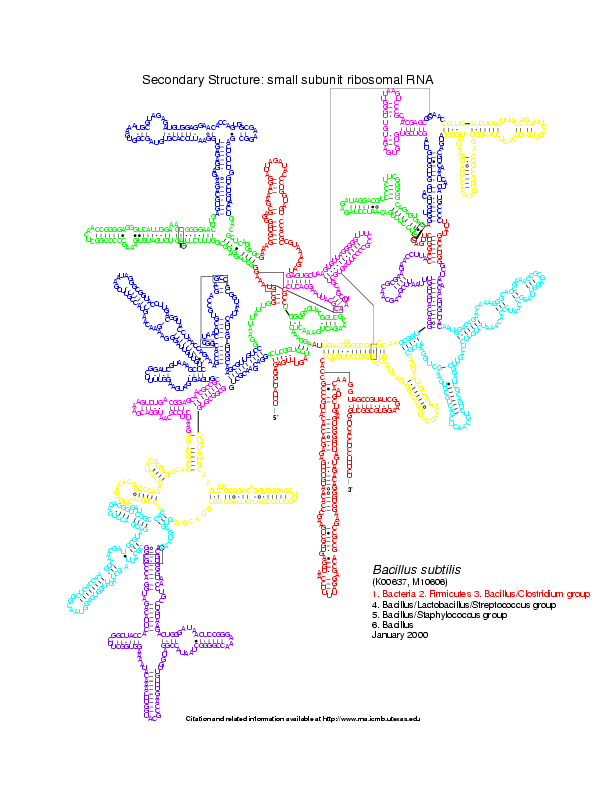
Level 1: the stems network comparison:
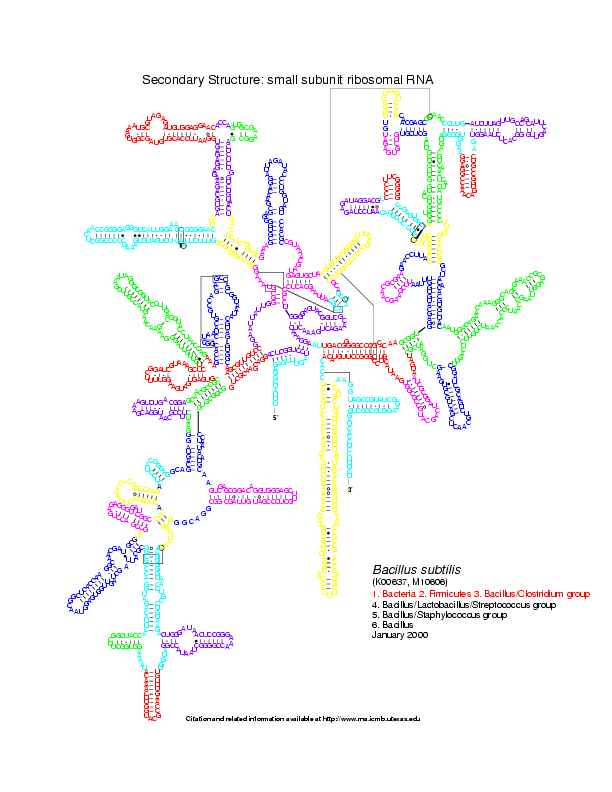

Level 2: the helices network comparison:
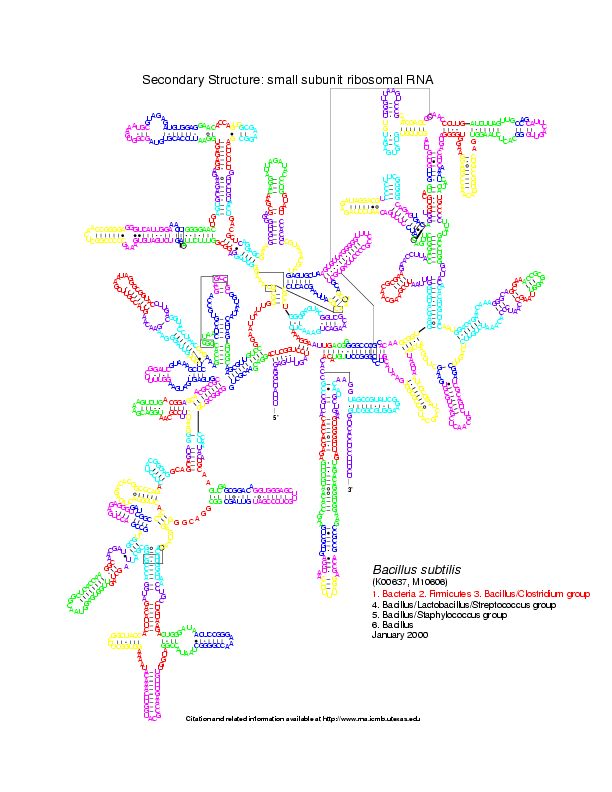

Note: you can view all these images in a flash animation using the migal web site
Indicate pseudo-knot only once
rnaconverter is used to convert RNA secondary structures
into the MiGaL xml file format. To convert the file
''d.16.b.B.subtilis.bpseq'' into xml, use rnaconverter -f bpseq
d.16.b.B.subtilis.bpseq -i -o d.16.b.B.subtilis.xml . You can
also use rnaconverter to encode a structure into the dot/parenthesis
file format with the option -d
Remark that the output of rnaconverter is a file without pseudo-knots. So, running migal on the xml files will not ask for pseudo-knots.
Dealing with nucleotide links
By default, the nucleotide links can only match nucleotide links and unpaired nucleotides can only match unpaired nucleotides. Mainly this is due to the set of edit operation used to compare the last level.
One solution is using the same coding as the one used in RNAforester with the option --forester
You can clearly see the difference with/without this option by consulting the following comparisons of the Introns Group I: with the --forester option and without the option. In particular take attention to the hairpin loops at the top right of the structures in the last level.