Julien Allali
MiGaL Tutorial
MiGaL Tutorial: Example 3, build phylogenetic trees using UPGMA method
This example explains how to build a phylogenetic tree
for a set of RNA secondary structures using phylo.py.
This python script is available in the directory tools in
the migal sources.
Algorithm description
phylo.py starts by computing the scoring
matrices (pairwise comparisons). Then the best score is selected. The
two
species corresponding to this score are removed from the matrix and
replaced by a consensus structure (built with migal -M -x ...).
Then the
scores between the new structure and other structures are cumputed and
so on until the matrix contains only one structure.
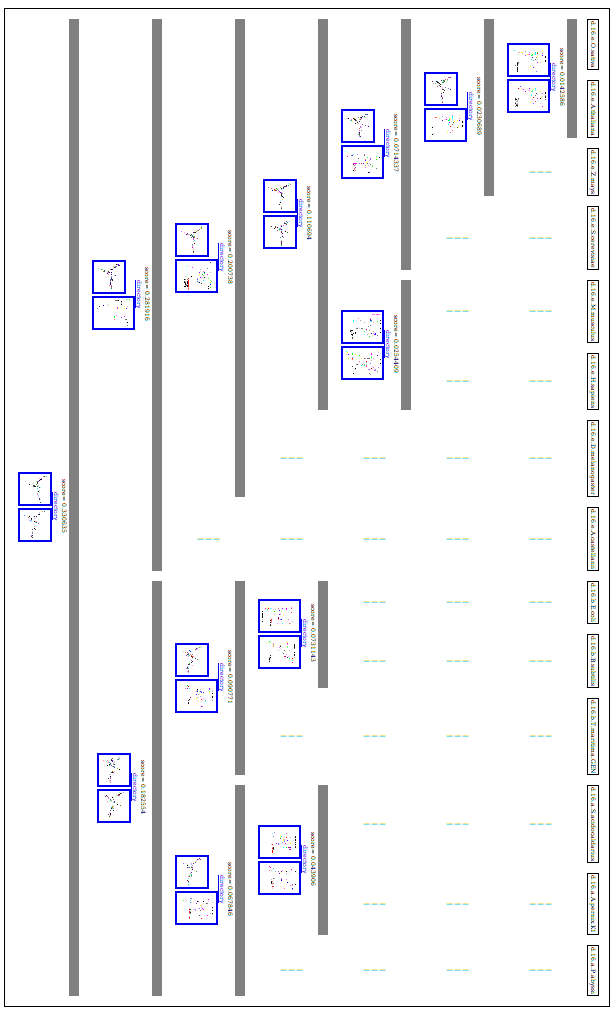
At each step i, a directory that represents the selected score in the matrix is created. For example, if we have three species A, B and C, we first compute the score between A and B, A and C, B and C. If the score for AC is the minimum, a directory 0_A_C is created and the comparison between A and C is re-run in that directory.
Concret example
The options for this script are the same as for the phylo_nj.py
script (see example2). In particular,
--html-rel and --html-abs allows one to
create
an html file that represents the computed tree with links to each
directory.
Using the same data of the previous example (8 16S structures + 2
postscripts)
and the command phylo.py --html-rel --phylip --rnaplot *xml,
we
obtain (in less than 1 minute):
- the phylogenetic tree in file intree
- a postscript representation of this tree in tree.ps
- four directories that represent hypothetic ancestors
- a html file that contains the tree with links to images, scores and alignments
Below is the file tree.ps

Second example:
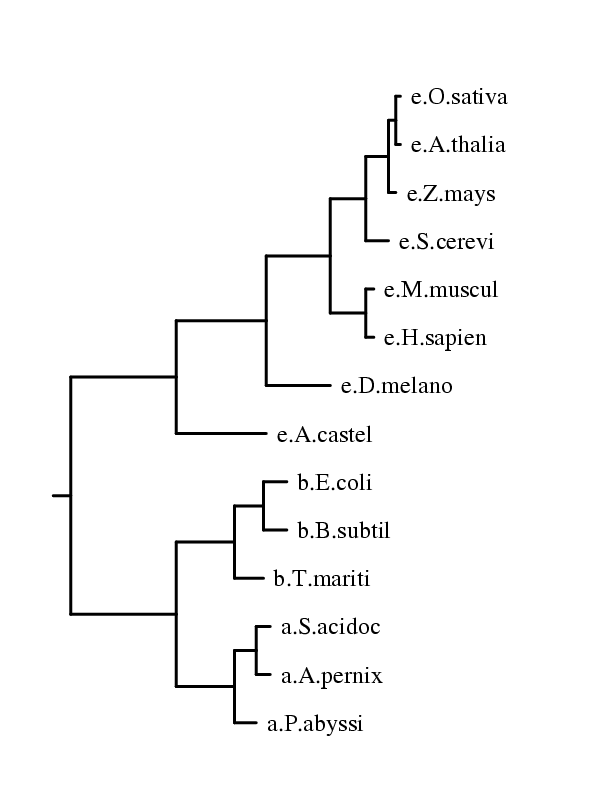
The second example is a tree built for 14 16S taken from the The Comparative RNA Web Site.
RNAs and postscripts are in this file. We
the run the following command to remove pseudo-knots:
for i in *bpseq
do
rnaconverter -i $i -o `basename $i bpseq`xml
done
and then phylo.py --html-rel --phylip --rnaplot *xml
(take about 2 minutes).
The tree drawn with phylip is shown below:
The html file of the tree is available here(screen-shot below):